Lupine Publishers | Trends in Ophthalmology Open Access Journal
Abstract
Background: Advances in genomics continue to enable the discovery of gene variants which cause various inherited ophthalmic disorders. Several case reports have shown an association between keratoconus and retinal disease but whether there is a genetic basis for this is still not known.
Methods: Clinical case study with Pentacam imaging, fundus autofluorescence (FAF), macular optical coherence tomography (OCT), electrophysiology studies, and genetic analysis.
Results: We report three brothers, two of whom have keratoconus and one who was found to have bilateral cone-rod dystrophy. This was supported by color vision and electrophysiology testing, fundus autofluorescence, and macular OCT findings. Genomic data analysis revealed three rare gene variants (MAP3K19, ADGRV1, and PIK3CG) common to the brother with cone-rod dystrophy and one brother with keratoconus. There was also a very significant variant in the CHST6 gene in the latter. Whole exome sequencing data revealed a rare missense variant for IMPG2 gene in both brothers.
Conclusion: Among the four genes with shared mutations in two of the brothers, IMPG2 has been linked to retinal disease while MAP3K19 and PIK3CG carry high risk scores for keratoconus pathogenesis. A highly damaging CHST6 variant detected in the brother with keratoconus is known to cause macular corneal dystrophy and corneal thinning. This study offers the first familial genetic analysis for keratoconus and cone-rod dystrophy. More studies with genomic investigations are needed in order to further elucidate the possible relationship between these two diseases.
Keywords: keratoconus; Cone-rod dystrophy; Autofluorescence; Electrophysiology; Genetic testing
Introduction
Keratoconus is a multifactorial degenerative corneal disorder characterized by corneal thinning and ectasia, with most cases occurring sporadically. However, it also occurs in families and may be inherited in an autosomal dominant or recessive pattern. By taking its pathophysiology into consideration, a number of candidate genes for keratoconus has been suggested, most notable of which are VSX1, ZNF469, SOD1 and miR184 [1-3]. The gene most concretely associated with keratoconus, albeit in a very small number of cases, is miR184 [4]. However, the evidence so far has been inconclusive regarding the degree of contribution of these genes to the development of disease.
Rarely, keratoconus has been reported in conjunction with various retinal disorders, including retinitis pigmentosa, optic disc pit, optic disc coloboma, and congenital Leber amaurosis [5-8]. The co-existence of diffuse tapetoretinal degeneration with keratoconus has also been described [9]. Cone and cone-rod dystrophies, a group of inherited retinal disorders, constitute other rare co-morbidities of keratoconus. The earliest report of cone dystrophy associated with keratoconus was made in 1995 by Wilhelmus, and four more cases of cone dystrophy associated with keratoconus have been reported since [10-13]. However, genetic analysis was not done in these cases, and whether a basis exists for the association of keratoconus with cone dystrophy is still not known.
Case Report
We report three brothers, two of whom were diagnosed with keratoconus and one who was found to have bilateral bull’s-eye maculopathy secondary to cone-rod dystrophy. The eldest and the youngest consulted at ages 53 and 45, respectively, for progressive blurring of vision. The eldest brother, P.V., had undergone penetrating keratoplasties for keratoconus in both eyes. Visual acuities were 8/10 and 6/10 in the right and left eyes. The 4-year old graft in the right eye was clear while there was mild haze in the older 24-year old graft of the other eye. Fundoscopy and macular optical coherence tomography (OCT) findings were unremarkable. We performed deep anterior lamellar keratoplasty on the left eye, and the graft did well for six years. Descemet stripping automated endothelial keratoplasty and phacoemulsification with toric intraocular lens implantation were done recently due to failing endothelium and a beginning cataract.
The youngest brother, G.V., presented with clear corneas bilaterally. Visual acuities were 7/10 on the right and 6/10 on the left eyes. An epiretinal membrane without traction was seen on macular OCT on the right. The left eye was diagnosed with keratoconus and collagen crosslinking was done. His Pentacam maps ten years later (Figure 1) show only mild posterior elevation in the right eye, while the left eye was stable with K2 51.2D and thinnest pachymetry of 488um.
The second brother, Z.V., consulted us six years after his brothers at age 50 for blurring of central vision described as a positive scotoma after an accident two years earlier. He also developed an epileptic disorder which necessitated multidrug maintenance treatment including levetiracetam and lamotrigine. He claimed to have always had good vision until then. The patient did not have regular intake of chloroquine or hydroxychloroquine. Family ocular history was negative for keratoconus or any other disorder. There was no evidence of nystagmus, and only a minor inferior stromal scar was found on slitlamp examination. Visual acuity was 3/10on the right eye and 4/10 on the left. Pentacam imaging (Figure 2) showed asymmetric with-the-rule bowtie astigmatism on the right due to the corneal scar, while the left eye had symmetric oblique bowtie astigmatism. Although the corneas were relatively thin, the elevation maps appeared normal and Belin-Ambrosio Enhanced Ectasia showed no signs of keratoconus in both eyes.
‘Bull’s-eye’ annular macular lesions were seen in both eyes on fundoscopic exam (Figure 3A) and testing with Hardy-RandRittler’s pseudoisochromatic plates (4th edn.) revealed severe redgreen color deficiency. Fundus autofluorescence (FAF) (Figure 3B) showed a hyperautofluorescent ring surrounding an area of hypo autofluorescence in the fovea of both eyes.
Spectral domain OCT (Figure 4) shows disruption of the outer layers of the fovea above the retinal pigment epithelium complex, specifically the external limiting membrane, the inner segmentouter segment junction or ellipsoid zone, and the cone outer segment tips line or interdigitation zone in both eyes. Follow-up after three years showed a decrease in BCVA to 1/20 on the right eye and 1/10 on the left. Repeat FAF showed an increase in the size of the macular lesions as well as increased hypo autofluorescence.
Electrophysiology
Electrophysiology tests were performed on Z.V. according to the International Society for Clinical Electrophyisiology of Vision (ISCEV) protocols. Pattern and Flash visual evoked potentials (PVEPFVEP) were severely subnormal, while Pattern electroretinogram (PERG) was undetectable in both eyes.
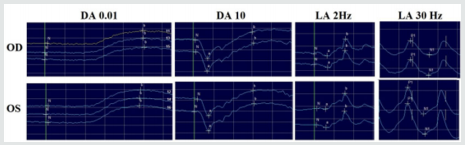
Electroretinogram (ERG) (Figure 5) was performed with DTL electrodes using complementary extra flashes apart from the ISCEV standard protocols in dark-adapted (DA) and light-adapted (LA) conditions. Scotopic ERG was normal, apart from a slight delay in the β-wave at DA 10 flash. Photopic ERG was again normal except for borderline implicit times in LA 30 Hz flicker flash. These results show a diagnosis of macular dystrophy with borderline recordings in photopic and scotopic ERG, suggestive of a cone-rod dystrophy.
Genetic Analysis
Z.V. and P.V., diagnosed with cone-rod dystrophy and bilateral keratoconus, respectively, underwent genomic data analysis as a family case study by Avellino Laboratories, Inc. (CA, USA), which specializes in corneal diseases. The focus was on genetic variants which were common to both brothers.
Using the Avellino keratoconus detection panel, Z.V. was found to have a total of six gene variants, three of which are shared with his brother (MAP3K19, ADGRV1, and PIK3CG), highlighted blue in Table 1. P.V. was found to have five gene variants, almost all of which have high risk scores for keratoconus pathogenesis. As mentioned previously, three of these variants, found in three different genes, are shared with his brother. PXN gene variants were also found in both brothers, but on different exons, highlighted yellow in Table 1. Additionally, P.V. has a very significant variant in CHST6 gene.
Because of severe pathogenic phenotype in the two brothers and possible diagnosis of cone-rod dystrophy, each individual’s variant analysis resulting from whole exome sequencing data was examined for other significant variants outside the keratoconus panel. A missense variant for IMPG2 gene in both brothers was detected (Table 2). IMPG1 gene however was not part of the panel.
Discussion
A diagnosis of cone-rod dystrophy in Z.V. was supported by color vision and electrophysiology testing, fundus autofluorescence, and macular OCT findings. Deteriorating vision and larger lesions on repeat FAF showed that the disorder was progressive. A drug-induced effect was ruled out because his anti-epileptic drugs levetiracetam and lamotrigine are not associated with any maculopathy [14].
Genomic data analysis with the Avellino detection panels revealed three mutations which carry high risk scores for keratoconus in Z.V. and four in P.V. Among these, PIK3CG and MAP3K19 are shared while PXN gene variants were found on different exons (Table 1). The fourth high-risk variant in P.V. is CHST6, which is associated with macular corneal dystrophy and corneal thinning. In a study by Dudakova et al., patients with this variant exhibited diffuse corneal thinning with paracentral steepening of the anterior corneal surface on Pentacam. This was graded as keratoconus by the software, but posterior corneal surface ectasia and focal thinning were not seen [15]. Among all the detected variants, this has the highest risk score for keratoconus pathogenesis and is the most deleterious and damaging.
Phosphatidylinositol-4,5-Bisphosphate 3-Kinase Catalytic Subunit Gamma (PIK3CG)
PIK3CG activates signaling cascades involved in cell growth, survival, proliferation, motility and morphology, and is involved in various immune, inflammatory and allergic responses. Mutations in this gene could lead to multiple phenotypes and secondary diseases and are seen in less than 1% of the population. Variant in this gene is mostly missense, which corresponds to amino acid changes of arginine to histidine [16]. PIK3CG gene variant has been reported in two other keratoconus cases in the Avellino archive, but not in any sample from control individuals. A search of the NCBI CLINGEN database shows that two cysteine deletion variants in the PIK3CG gene have been reported by the Institute of Human Genetics of the Polish Academy of Sciences to be linked to keratoconus, although the significance is still considered uncertain [17].
Mitogen-Activated Protein Kinase Kinase Kinase 19 (MAP3K19)
Downstream signaling pathways of MAP3 kinases induce proliferation, differentiation, inflammation and eventually apoptosis. MAP3K19 variant found in both brothers is also a missense mutation, where serine is replaced by leucine [18-19]. This is also a rare mutation in the population (< 1%), and has been reported in seven other keratoconus cases in the Avellino archive but not in any of control samples. Based on predictive computational algorithms, this variant found in both brothers is significant and carries a high-risk score.
Adhesion G Protein-Coupled Receptor V1 (ADGRV1)
ADGRV1 is a cell-surface protein, a G-protein coupled receptor, which has an essential role in the development of hearing and vision [20-23]. The variant found in both brothers is a missense variant located at a Calx-beta domain. This is also a rare mutation seen in less than 1% of the population and has been reported in three other keratoconus cases in the Avellino archive. However, it has also been seen in one sample from control individuals; this possibly damaging variant therefore does not carry a risk score.
Interphotoreceptor Matrix Proteoglycan 2 (IMPG2)
MPG1/2 genes are very well studied in ocular
pathogenesis, and there are several studies on IMPG1- and IMPG2-associated
retinitis pigmentosa, vitelliform macular dystrophy, and other ocular diseases.
This missense variant is significant in that it affects DNA replication,
nuclear organization and gene transcription [24]. The population allele
frequency is very small, suggesting that this variant is a very rare mutation
in total population as well. In summary, among the shared gene variants, IMPG2
is linked to retinal disease while both MAP3K19 and PIK3CG carry high risk
scores for keratoconus pathogenesis. This study offers the first familial
genetic analysis for keratoconus and cone-rod dystrophy. However, more studies
with genomic investigations are needed in order to further elucidate the
possible relationship between these two diseases.
https://lupinepublishers.com/ophthalmology-journal/fulltext/keratoconus-and-cone-rod-dystrophy-among-brothers-clinical-case-study-and-genetic-analysis.ID.000133.php
For more Lupine Publishers Open Access Journals Please visit our website: https://twitter.com/lupine_online
For more Trends in Ophthalmology Please Click
Here: https://lupinepublishers.com/ophthalmology-journal/
To Know more Open Access Publishers Click on Lupine Publishers
Follow on Linkedin : https://www.linkedin.com/company/lupinepublishers
Follow on Twitter : https://twitter.com/lupine_online

No comments:
Post a Comment